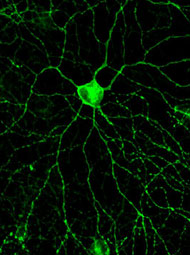
Quando os neurônios visuais degeneram no olho, não há tratamento possível. Mas um novo estudo traz um pouco de esperança. Ratos injetados com uma proteína receptora de luz encontrada em algas parecem recuperar alguma habilidade de processar informações visuais.
Os fotoreceptores convertem a luz em impulsos elétricos que são enviados ao cérebro através dos neurônios da retina. Se esses fotoreceptores forem danificados ou destruídos-como acontece, por exemplo, na retinite pigmentosa- os neurônios retinianos ficam sem sinais para transmitir, e a visão é perdida. Zhuo-Hua Pan, um neurocientista especializado em visão, que trabalha na Wayne State University em Detroit, Michigan, começou a se perguntar se não haveria uma maneira de fazer com que os neurônios retinianos agissem mais como fotoreceptores.
Um gene de foto-pigmento de algas, recém-clonado, pareceu a Pan bom ponto de partida. O foto-pigmento permite que as algas detectem luz, e como ele é codificado por um único gene, é fácil incorporar o DNA a um vírus. Pan e seus colegas injetaram o vírus modificado nos olhos de ratos que não tinham fotoreceptores na retina. Três a quatro semanas após a injeção, o marcador fluorescente ligado ao gene coloriu os neurônios retinianos, indicando que eles estavam expressando o foto-pigmento. Normalmente, neurônios retinianos não transmitem sinais ao córtex visual em resposta à luz, mas estes neurônios o fizeram. E o efeito foi duradouro: pelo menos 12 horas após a injeção, os neurônios retinianos continuaram a disparar em resposta à luz, segundo relato de Pan em artigo publicado na revista Neuron de 6 de abril de 2006.
Será que esses sinais podem produzir visão útil? Pan e seus colegas ainda não sabem, pois eles ainda precisam fazer estudos comportamentais dos ratos, com a finalidade de testar a sua acuidade visual.
Resumo do trabalho publicado:
Ectopic Expression of a Microbial-Type Rhodopsin Restores Visual Responses in Mice with Photoreceptor Degeneration
Anding Bi, Jinjuan Cui, Yu-Ping Ma, Elena Olshevskaya, Mingliang Pu, Alexander M. Dizhoor and Zhuo-Hua Pan
The death of photoreceptor cells caused by retinal degenerative diseases often results in a complete loss of retinal responses to light. We explore the feasibility of converting inner retinal neurons to photosensitive cells as a possible strategy for imparting light sensitivity to retinas lacking rods and cones. Using delivery by an adeno-associated viral vector, here, we show that long-term expression of a microbial-type rhodopsin, channelrhodopsin-2 (ChR2), can be achieved in rodent inner retinal neurons in vivo. Furthermore, we demonstrate that expression of ChR2 in surviving inner retinal neurons of a mouse with photoreceptor degeneration can restore the ability of the retina to encode light signals and transmit the light signals to the visual cortex. Thus, expression of microbial-type channelrhodopsins, such as ChR2, in surviving inner retinal neurons is a potential strategy for the restoration of vision after rod and cone degeneration.
Esta é, sem dúvida, mais uma esperança para os portadores de retinite pigmentosa e outras doenças semelhantes.
Link para o artigo completo no site da revista Neuron

 Graças à pesquisa com os "super-ratos", a vida das pessoas que sofrem de distrofias musculares e outras doenças semelhantes pode mudar. Estão a caminho dois novos tratamentos que bloqueiam uma proteína chamada miostatina, que normalmente retarda o crescimento dos músculos.
Graças à pesquisa com os "super-ratos", a vida das pessoas que sofrem de distrofias musculares e outras doenças semelhantes pode mudar. Estão a caminho dois novos tratamentos que bloqueiam uma proteína chamada miostatina, que normalmente retarda o crescimento dos músculos.